- COORDINATION (chimie) - Composés de coordination
- COORDINATION (chimie) - Composés de coordinationUn composé de coordination est une entité chimique constituée d’un atome central et d’un ou plusieurs coordinats, ou ligands, éventuellement en nombre supérieur à celui qui résulterait de la valence ou du degré d’oxydation usuel de l’atome central. Un certain nombre de ces composés avaient été décrits dès le milieu du XVIIIe siècle; dans la seconde moitié du XIXe siècle, divers chimistes, en particulier Blumstrand et S. M. Jorgensen, tentèrent d’appliquer à ce type de composés, appelés alors improprement «combinaisons moléculaires», les théories de la valence et de l’enchaînement des atomes (on dit maintenant «caténation») qui venaient de faire preuve de leur bien-fondé dans le domaine de la chimie organique. L’idée de la coordination d’ions ou de groupes d’atomes suivant une disposition géométrique autour d’un ion central fut émise en 1893 par Alfred Werner, véritable fondateur de la chimie de coordination, qui reçut le prix Nobel en 1913. Werner montra, par exemple, que les propriétés des complexes ayant un nombre de coordination égal à 6 pouvaient s’interpréter en admettant que les six groupements fixés étaient disposés, autour de l’atome central, aux sommets d’un octaèdre régulier. Ainsi, la combinaison moléculaire CoI3,6NH3 devait en réalité se formuler [Co(NH3)6]I3. Une autre propriété importante que Werner put expliquer ainsi était l’existence des isomères cis-trans : plus tard, il dédoubla des racémiques dont il avait prévu l’existence.En France, à partir de 1906, Delépine compléta l’œuvre de Werner, que les travaux de Miolati et Rosenheim étendirent aux cas des oxyanions polynucléaires dérivant des isopolyacides et des hétéropolyacides fréquemment rencontrés dans le cas du molybdène (VI) et du tungstène (VI) mais aussi à un degré moindre avec le vanadium (V), le niobium (V), le tantale (V) et l’uranium (VI). Jusqu’au milieu du XXe siècle, divers physico-chimistes tels que Jander, Keggin, Pauling, Souchay réussirent à adapter les conceptions de Werner à ces composés.Par ailleurs, après les premières théories ioniques et électroniques de Kossel, Lewis, Langmuir, Sidgwick, il fallut attendre 1930-1940 pour que des concepts plus élaborés concernant la liaison de coordination apparaissent avec la théorie de l’hybridation des orbitales de Pauling, Prix Nobel de Chimie en 1954, et de Slater. Avec l’accumulation de données expérimentales ne corroborant pas cette théorie, notamment en ce qui concerne les propriétés magnétiques de nombreux complexes métalliques, il fallut avoir recours, vers 1950, sous l’impulsion de Balhausen, Cotton, Griffith, C. K. Jorgensen et Orgel, à la théorie du champ cristallin (en anglais crystal field theory , C.F.T.), ultérieurement étendue et généralisée par la théorie du champ de ligand (en anglais ligand field theory , L.F.T.).Des milliers de complexes sont aujourd’hui connus, et un très grand nombre d’entre eux ont des applications pratiques décisives dans certains domaines tels que celui de la catalyse homogène (cf. COMPLEXES - Chimie). Il faut noter que les recherches relatives aux composés de coordination se sont constamment développées. C’est ainsi, que soixante-quatorze ans après Werner, ce sont trois chimistes ayant synthétisé de nouveaux ligands tels que les molécules creuses du type éther-couronne ou cryptate qui se voient attribuer le prix Nobel de chimie en 1987: Cram, Lehn et Pedersen.1. La coordinenceLa plupart des complexes métalliques comprennent un atome métallique constituant l’atome central et des ions, des atomes ou des molécules coordinés. L’atome métallique central est généralement à l’état de cation avec un degré d’oxydation positif bien que l’on connaisse des complexes où le degré d’oxydation formel du métal est nul, comme dans Ni(CO)4, voire négatif comme pour [Fe(CO)4]2-. Werner ayant effectué ses travaux bien antérieurement à l’énoncé de la théorie électronique des atomes admit que l’atome central disposait, d’une part, de valences principales qui lui permettaient de fixer des atomes et des radicaux et, d’autre part, de valences secondaires qui retenaient des molécules telles que H2O ou NH3: la somme de ces deux nombres constitue alors le nombre de coordination. Job appelle coordinence ce nombre de coordination et il considère qu’il s’agit d’un invariant prenant le plus souvent les valeurs 4 ou 6 pour des familles de complexes d’un même cation métallique.La charge électrique du complexe est égale à la somme algébrique du degré d’oxydation ou de l’électrovalence de l’atome central et de ceux des atomes et des ions coordinés. On peut ainsi obtenir des anions complexes du type [FeIII(CN)6]3-, où le fer est au degré d’oxydation + 3, des cations complexes du type [CrIII(H2O)6]3+ ou des composés moléculaires à charge nulle comme [PtIICl2(NH3)2].La théorie a été étendue sans difficulté à des ions complexes dont l’atome central est un non-métal: [S4]2-, [Si6]2-, [NR4]+, etc. Elle a aussi été appliquée avec succès aux complexes polynucléaires qui se forment lorsque les coordinats peuvent jouer le rôle de pont entre les ions centraux; comme c’est le cas pour le nitrate de décammine- 猪-amido-dicobalt (III), [(NH3)5Co 漣NH2 漣Co(NH3)5](NO3)5, découvert par Werner en 1908.De nos jours, certaines de ces notions ont également évolué: c’est ainsi que l’on connaît des composés de coordination où le nombre de coordination prend les valeurs 2, 3, 4, 5, 6, 7, 8, voire 9 à 12: le complexe K4[Th(C24)4(H2O)2],2H2O du thorium (IV) avec les ions oxalate C242- présente un nombre de coordination de 10.Cependant, dans le cas de nombres de coordination élevés il apparaît fréquemment des structures à géométrie distordue. Sur l’exemple du complexe du thorium (IV), on peut noter qu’il faut bien tenir compte de la présence de coordinats multidentés (ici C242- est un coordinat bidenté) pour définir correctement le nombre de coordination.2. La liaison de coordinenceThéorie ioniqueIntroduite par Kossel, la théorie ionique ne sera pas développée ici, car il est évidemment peu réaliste de considérer que les interactions entre un cation métallique et des coordinats puissent être purement ioniques.Théorie électroniqueL’hypothèse de base proposée par Sidgwick consiste à considérer, dans le cas d’un complexe octaédrique, l’établissement de six liaisons datives entre le cation métallique central jouant le rôle d’accepteur d’électrons, c’est-à-dire d’acide de Lewis, et six doublets électroniques de ligand(s) jouant un rôle de donneur d’électrons, c’est-à-dire de base de Lewis. Les liaisons orientées dans l’espace permettent d’expliquer les stéréo-isoméries. Sidgwick a introduit la notion de nombre atomique effectif qui représente le nombre total des électrons entourant l’atome central compte tenu des doublets mis en commun; si Z est le numéro atomique de l’atome central, V la valeur algébrique de la valence de l’ion correspondant, et C la coordinence, on a: Z eff = Z 漣 V + 2C . Par exemple, pour le complexe Ni(CO)4: Z = 28, V = 0, C = 4 et Z eff = 36; le nombre atomique effectif correspondant alors au numéro atomique du gaz rare situé à la fin de la période considérée, c’est-à-dire le krypton dans l’exemple choisi. Si l’on ne considère que les 10 électrons périphériques 3d et 4s du nickel (0), son environnement est alors un environnement à 18 électrons. On a souvent postulé que les complexes satisfaisant à cette règle dite du gaz rare, ou règle des 18 électrons, étaient dotés d’une stabilité particulière, et on a souvent utilisé cette règle pour déterminer les stœchiométries des composés de coordination. Là encore, les données récentes montrent que, si cette règle s’applique strictement dans le cas de ligands possédant des orbitales 兀 telles que CO, C-, etc., elle ne peut être appliquée dans le cas général des métaux de la première série de transition où l’environnement électronique peut varier de 12 à 22 (toujours dans le cas de complexes octaédriques) et qu’il existe de nombreuses exceptions pour les métaux des deuxième et troisième séries de transition où, cependant, l’environnement électronique ne dépasse jamais 18.Théorie des liaisons de valenceDans le cas de la théorie des liaisons de valence, Pauling et Slater ont montré que, pour un complexe octaédrique, on pouvait faire appel à la notion d’hybridation de deux orbitales n d, d’une orbitale (n + 1)s et de trois orbitales (n + 1)p pour aboutir à la formation de six orbitales hybrides d2sp3 équivalentes occupées par les paires électroniques des coordinats. Un raisonnement analogue pourrait être effectué pour un complexe carré plan (hybridation dsp2) ou pour une symétrie tétraédrique (hybridation sp3).La figure 1 illustre cette démarche. On constate que, si l’ion Fe3 possède bien 5 électrons non appariés, l’ion complexe [Fe(CN)6]3- n’en possède plus qu’un puisque deux orbitales d sont occupées par les électrons des ligands. Le moment magnétique d’un complexe étant donné par la formule simplifiée 猪 = 連n (n + 2) M.B., où M.B. représente le magnéton de Bohr et n le nombre d’électrons «célibataires», cela signifie que tous les complexes d’un même cation métallique devraient posséder des propriétés magnétiques voisines, ce qui s’est avéré très rapidement faux puisque, par exemple, un complexe comme [Co6]3- est paramagnétique, alors que [Co(NH3)6]3+ est diamagnétique. Seules les théories C.F.T. et L.F.T. peuvent expliquer les lacunes des théories précédentes.Théorie électrostatique du champ cristallinLes cinq orbitales d des ions des métaux de transition possèdent toutes la même énergie dans l’ion libre: elles sont dégénérées. Lorsque l’ion métallique se trouve soumis au champ électrostatique des doublets électroniques qui assurent la fixation des coordinats, il y a levée de dégénérescence et séparation des niveaux d’orbitales d. L’effet produit sur les énergies de ces orbitales dépend de la configuration géométrique du complexe. Si l’on considère, par exemple, le cas d’un complexe octaédrique, il est clair que les lobes des trois orbitales dxy , dyz , dzx étant centrés suivant les bissectrices des axes de coordonnées, les effets de répulsion entre ces orbitales et celles des coordinats centrées sur les axes seront minimaux, alors que, dans le cas des deux orbitales dz 2 et dx 2-y 2, ces répulsions seront maximales; d’où deux niveaux énergétiques séparés par une valeur 0 dans l’échelle des énergies. 0 croît avec le degré d’oxydation du cation métallique central et aussi lorsque, pour une colonne donnée de la classification périodique, on passe de la première à la deuxième et enfin à la troisième série des métaux de transition. Lorsque 0 n’est pas trop grand, la répartition des électrons de l’ion central suit la règle de Hund et fournit le maximum d’électrons célibataires: on a un complexe «à champ faible» ou «à haut spin». Quand 0 est suffisamment grand, les électrons occupent d’abord les orbitales de plus basse énergie (dxy , dyz , dzx ) jusqu’à saturation: on a un complexe «à champ fort» ou «à bas spin». Pour un ion central déterminé, on peut ranger les coordinats dans l’ordre croissant des 0 qu’ils provoquent (série spectrochimique): iodure 麗 bromure 麗 chlorure 麗 fluorure 麗 hydroxyde 麗 oxalate 麗 eau 麗 pyridine 麗 ammoniac 麗 éthylènediamine 麗 bipyridyle.Cet ordre est indépendant de la nature de l’ion métallique central; pour un cation donné, il existe une valeur limite de 0 à partir de laquelle, lorsque le champ de coordinat croît, la règle de Hund n’est plus respectée; il y a passage des complexes à «haut spin» aux complexes à «bas spin».Cette théorie peut être généralisée à d’autres structures régulières, comme on le voit sur le schéma de principe de la figure 2.La théorie permet aussi de rendre compte qualitativement de l’effet Jahn-Teller qui est une déformation du polyèdre régulier considéré, même lorsque les coordinats sont identiques. C’est ainsi que l’ion Cu2+ (d 9) peut adopter dans une structure octaédrique la configuration électronique t62g eg 3 avec une répartition de deux électrons dans l’orbitale dz 2 et d’un seul électron dans l’orbitale dx 2-y 2. Il en résulte un «effet d’écran» maximal suivant l’axe z et donc une distorsion de l’octaèdre par élongation. C’est aussi le cas des ions d4 à haut spin (Cr2+, Mn3+) où l’octaèdre est déformé soit par élongation (densité électronique maximale dans dz 2), soit par aplatissement (densité électronique maximale dans dx 2-y 2).Théorie du champ de coordinatsLa notion d’orbitales moléculaires permet l’approche la plus complète et la plus rationnelle de la liaison de coordination. En effet, la théorie du champ cristallin est fondée sur l’hypothèse peu réaliste de coordinats assimilés à des charges ponctuelles alors que leurs orbitales se recouvrent avec celles de l’ion métallique. Les orbitales moléculaires peuvent être obtenues suivant différents modèles théoriques. La méthode L.C.A.O. (linear combination of atomic orbitals ) consiste à trouver des combinaisons linéaires appropriées des orbitales de l’ion central et des orbitales des coordinats. Dans le cas des complexes octaédriques d’un ion métallique de la première série de transition, six orbitales, de l’ion métallique (3dz 2, 3dx 2-y 2, 4s, 4px , 4py , 4pz ) possèdent des symétries permettant la formation de liaisons 靖 avec les coordinats et trois d’entre elles (3dxy , 3dyz , 3dzx ) sont disponibles pour former des liaisons 神. Les cinq orbitales 3d de l’ion métallique sont donc encore séparées en deux groupes: Eg , antiliantes, et 2g , non liantes (fig. 3). Ce type de raisonnement, généralisé aux cas plus complexes des cations lourds des deuxième et troisième séries des métaux de transition, permet l’interprétation des caractéristiques des complexes métalliques, notamment des propriétés magnétiques et spectroscopiques (transitions dd et bandes de transfert de charge), ainsi que de la stabilisation due au champ de coordinats. Cette dernière, calculée suivant l’occupation des orbitales Eg et 2g , est nulle pour les complexes octaédriques à haut spin des ions de structure électronique d0, d5 et d10 (Ca2+, Mn2+ et Zn2+) et maximale pour les ions de structure d3 et d8 (V2+ et Ni2+).3. IsomériesCertaines isoméries ne font pas intervenir la stéréostructure des complexes: isomérie de coordination , par exemple pour
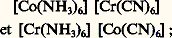 isomérie de position qui est une isomérie de coordination à l’intérieur d’un ion complexe polynucléaire; isomérie d’ionisation illustrée par [Co(NH3)5(SO4)]Br et [Co(NH3)5Br] [S4]; isomérie d’hydratation , lorsqu’un même nombre de molécules d’eau se répartit de manière différente à l’intérieur et à l’extérieur de l’ion complexe, ce qui est le cas pour [Cr(H2O)6]Cl3, [CrCl(H2O)5]Cl2, H2O et [CrCl2(H2O)4]Cl, 2H2O; isomérie d’un groupe donneur : n -propylamine et isopropylamine par exemple; isomérie de sel qui fait intervenir deux groupements isomères coordinés différemment, tels que 漣2 et 漣O 漣NO; isomérie de valence que l’on rencontre
isomérie de position qui est une isomérie de coordination à l’intérieur d’un ion complexe polynucléaire; isomérie d’ionisation illustrée par [Co(NH3)5(SO4)]Br et [Co(NH3)5Br] [S4]; isomérie d’hydratation , lorsqu’un même nombre de molécules d’eau se répartit de manière différente à l’intérieur et à l’extérieur de l’ion complexe, ce qui est le cas pour [Cr(H2O)6]Cl3, [CrCl(H2O)5]Cl2, H2O et [CrCl2(H2O)4]Cl, 2H2O; isomérie d’un groupe donneur : n -propylamine et isopropylamine par exemple; isomérie de sel qui fait intervenir deux groupements isomères coordinés différemment, tels que 漣2 et 漣O 漣NO; isomérie de valence que l’on rencontre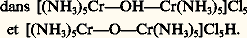 Les polyméries sont à rapprocher de ces isoméries, par exemple [Pt Cl2(NH3)2] et [Pt (NH3)4] [Pt Cl4]. Il y a, en outre, des stéréo-isoméries comme l’isomérie géométrique cis-trans des complexes tétracoordinés plans et l’isomérie optique , que l’on rencontre dans les complexes à structure tétraédrique, et dans les cas de divers complexes octaédriques tels que ceux représentés sur la figure 4.4. Stabilité et réactivitéLa formation d’un complexe en solution correspond au remplacement d’une ou de plusieurs molécules de solvant par un ou plusieurs coordinats A. Dans le cas de la fixation de plusieurs coordinats, la formation du complexe MAr se fait par étapes suivant des équilibres successifs du type MAr-1 + A 曆 MAr . La stabilité ionique du complexe MAr est alors chiffrée par les valeurs des constantes de stabilité ionique consécutives (ou successives): K r = [MAr ]/[MAr-1 ][A] ou celles des constantes de stabilité ionique globales (ou cumulatives): 廓 r = [MAr ]/[A]r ; 廓 r = K 1 . K 2 . K 3 ... K r (cf. COMPLEXES – Chimie). Il est très important de pouvoir prévoir a priori la stabilité d’un complexe donné. Parmi les différents facteurs influençant cette stabilité, on peut distinguer ceux qui sont liés à la nature de l’ion métallique central et ceux qui sont liés à la nature des coordinats.Ion métallique centralSi l’on considère qu’un simple modèle électrostatique rend compte des interactions cation-coordinat(s), les constantes de stabilité pour des ions de même charge sont inversement proportionnelles au rayon ionique de l’ion métallique: plus le rayon ionique est petit, plus le complexe est stable.En outre, les stabilités sont d’autant plus importantes que l’ion métallique central est chargé; c’est ainsi que l’ion Fe3+, plus petit et plus chargé, exerce une attraction plus forte que l’ion Fe2+, plus gros et de charge plus faible. Ces règles ne sont pas toujours respectées: pour les complexes de l’acide éthylènediamine-tétracétique (cf. COMPLEXES - Chimie) avec les métaux du groupe IIA de la classification périodique des éléments, la stabilité suit la séquence béryllium 麗 magnésium 麗 calcium 礪 strontium 礪 baryum; la stabilité des complexes du cuivre (I), argent (I) et or (I) croît fréquemment lorsque le rayon ionique augmente; des cations de même charge et de rayons ioniques voisins tels calcium Ca2+ (0,099 nm) et cadmium Cd2+ (0,097 nm) conduisent à des complexes de stabilités très différentes avec un coordinat donné. Ces contre-exemples montrent bien que d’autres facteurs doivent être envisagés.La structure électronique des ions métalliques joue un rôle déterminant. Pour les métaux bivalents, la série d’Irving et Williams est la plus connue: Mn 麗Fe 麗Co 麗Ni 麗Cu 礪Zn. Elle s’interprète classiquement à l’aide de la théorie du champ des coordinats et a été vérifiée rigoureusement (Irving, 1959).Il faut considérer simultanément la relation entre la nature des atomes donneurs et celle de l’ion métallique accepteur. C’est ainsi que les distinctions entre cations ont été discutées par Ahrland et al. (1958) et Schwarzenbach (1961):– classe a . Cations à configuration électronique externe de gaz nobles: Li+, Na+, K+, Be2+, Mg2+, Ca2+, Sr2+, Ba2+, Al3+... Ces ions possèdent une faible polarisabilité, une forte électropositivité, un faible rayon ionique, une charge positive élevée et forment des liaisons de type ionique. On aura donc l’ordre d’affinité suivant avec les principaux groupes donneurs:
Les polyméries sont à rapprocher de ces isoméries, par exemple [Pt Cl2(NH3)2] et [Pt (NH3)4] [Pt Cl4]. Il y a, en outre, des stéréo-isoméries comme l’isomérie géométrique cis-trans des complexes tétracoordinés plans et l’isomérie optique , que l’on rencontre dans les complexes à structure tétraédrique, et dans les cas de divers complexes octaédriques tels que ceux représentés sur la figure 4.4. Stabilité et réactivitéLa formation d’un complexe en solution correspond au remplacement d’une ou de plusieurs molécules de solvant par un ou plusieurs coordinats A. Dans le cas de la fixation de plusieurs coordinats, la formation du complexe MAr se fait par étapes suivant des équilibres successifs du type MAr-1 + A 曆 MAr . La stabilité ionique du complexe MAr est alors chiffrée par les valeurs des constantes de stabilité ionique consécutives (ou successives): K r = [MAr ]/[MAr-1 ][A] ou celles des constantes de stabilité ionique globales (ou cumulatives): 廓 r = [MAr ]/[A]r ; 廓 r = K 1 . K 2 . K 3 ... K r (cf. COMPLEXES – Chimie). Il est très important de pouvoir prévoir a priori la stabilité d’un complexe donné. Parmi les différents facteurs influençant cette stabilité, on peut distinguer ceux qui sont liés à la nature de l’ion métallique central et ceux qui sont liés à la nature des coordinats.Ion métallique centralSi l’on considère qu’un simple modèle électrostatique rend compte des interactions cation-coordinat(s), les constantes de stabilité pour des ions de même charge sont inversement proportionnelles au rayon ionique de l’ion métallique: plus le rayon ionique est petit, plus le complexe est stable.En outre, les stabilités sont d’autant plus importantes que l’ion métallique central est chargé; c’est ainsi que l’ion Fe3+, plus petit et plus chargé, exerce une attraction plus forte que l’ion Fe2+, plus gros et de charge plus faible. Ces règles ne sont pas toujours respectées: pour les complexes de l’acide éthylènediamine-tétracétique (cf. COMPLEXES - Chimie) avec les métaux du groupe IIA de la classification périodique des éléments, la stabilité suit la séquence béryllium 麗 magnésium 麗 calcium 礪 strontium 礪 baryum; la stabilité des complexes du cuivre (I), argent (I) et or (I) croît fréquemment lorsque le rayon ionique augmente; des cations de même charge et de rayons ioniques voisins tels calcium Ca2+ (0,099 nm) et cadmium Cd2+ (0,097 nm) conduisent à des complexes de stabilités très différentes avec un coordinat donné. Ces contre-exemples montrent bien que d’autres facteurs doivent être envisagés.La structure électronique des ions métalliques joue un rôle déterminant. Pour les métaux bivalents, la série d’Irving et Williams est la plus connue: Mn 麗Fe 麗Co 麗Ni 麗Cu 礪Zn. Elle s’interprète classiquement à l’aide de la théorie du champ des coordinats et a été vérifiée rigoureusement (Irving, 1959).Il faut considérer simultanément la relation entre la nature des atomes donneurs et celle de l’ion métallique accepteur. C’est ainsi que les distinctions entre cations ont été discutées par Ahrland et al. (1958) et Schwarzenbach (1961):– classe a . Cations à configuration électronique externe de gaz nobles: Li+, Na+, K+, Be2+, Mg2+, Ca2+, Sr2+, Ba2+, Al3+... Ces ions possèdent une faible polarisabilité, une forte électropositivité, un faible rayon ionique, une charge positive élevée et forment des liaisons de type ionique. On aura donc l’ordre d’affinité suivant avec les principaux groupes donneurs: – classe b . Cations à configuration électronique externe à dix-huit électrons: Cu+, Ag+, Au+, Zn2+, Cd2+, Hg2+... Les caractéristiques sont opposées à celles des ions de classe a, conduisant à des liaisons essentiellement covalentes, donc à des ordres d’affinité inversés.– classe c . De caractère intermédiaire, cette classe comprend notamment les ions des métaux de transition... Fe2+, Co2+, Ni2+, Cu2+.Ce type de notion a été développé par Pearson (1963) dans la théorie des acides (ions métalliques) durs et mous et des bases (coordinats) dures et molles. C’est la théorie H.S.A.B. (hard and soft acid base theory ). La classification correspondante présente de grandes analogies avec la précédente, la classe a représentant les acides durs et la classe b les acides mous. La règle fondamentale stipule que les acides durs préfèrent les bases dures et que les acides mous préfèrent les bases molles. C’est ainsi que les complexes fluorés de l’aluminium seront stables (F- base dure et Al3+ acide dur) alors que les complexes fluorés du mercure seront peu stables (Hg2+ acide mou).CoordinatsNature des atomes donneursDans la théorie H.S.A.B., les coordinats sont aussi classés en bases dures et molles. Une base dure aura d’une façon générale une faible polarisabilité, une grande électronégativité, une charge négative, une petite dimension et formera des liaisons ioniques. C’est le cas, par exemple, de H2O, OH-, -, P43-, S2-4 ... Les bases molles ont des caractéristiques inverses: R2S, RSH, RS-, I-, SC-, (S23)2-...Effets chélate et macrocycliqueLa stabilité des complexes est fortement accrue par chélation et par effet macrocyclique (cf. COMPLEXES - Chimie).Dimension et nombre de cyclesLes cycles pentagonaux les plus stables sont ceux qui ne renferment pas de double liaison (éthylènediamine...); les cycles hexagonaux les plus stables sont ceux qui contiennent une ou deux doubles liaisons ( 廓-dicétones, esters 廓-cétoniques).Plus le nombre de cycles formés est élevé, plus la stabilité est importante; la stabilité croît lorsque l’on passe de NH3 (0 cycle) à l’éthylènediamine (1 cycle), à la diéthylènetriamine (2 cycles), puis à la triéthylènetétramine (3 cycles).Propriétés acido-basiques du coordinatLes ions hydrogène et l’ion métallique sont des acides de Lewis, donc des accepteurs d’électrons. Il y a donc une similitude entre la complexation de l’ion métallique par un coordinat et la neutralisation d’une base par les ions hydrogène. En outre, la plupart des coordinats sont des bases conjuguées d’acides, et on peut penser que, d’une façon générale, plus le pK de la base est élevé, plus la stabilité du complexe métallique est grande. Cette corrélation est souvent vérifiée, mais là aussi il y a de nombreuses exceptions.Influence de substituant(s) sur le coordinatCes substitutions peuvent, d’une part, modifier les propriétés acido-basiques du coordinat, donc la stabilité des complexes, et, d’autre part, induire des effets stériques qui peuvent être importants et largement compenser les variations de stabilité dues aux variations de basicité.RésonancesLes résonances augmentent toujours la stabilité des complexes; pour qu’elles aient lieu, il faut que ceux-ci aient une structure plane ou s’écartant peu de la planéité; l’augmentation du domaine offert aux électrons délocalisés accroît encore la stabilité; c’est le cas des porphyrines ou, mieux encore, des phtalocyanines.Stabilisation de degrés d’oxydation inhabituelsLa coordination permet de stabiliser certains degrés d’oxydation inhabituels.Les degrés d’oxydation les plus petits, éventuellement même négatifs, sont stabilisés par coordination avec CO, C-, RCN, PCl3, la 2-2 -bispyridine; exemples: Na2[Fe(CO)4], qui contient Fe(face=F0019 漣 II), ou K4[Ni(CN)4] et Ni(CO)4, qui contiennent Ni(0). On attribue cette stabilisation à la formation d’orbitales moléculaires du type 神 entre le métal et son coordinat.Les degrés d’oxydation élevés sont stabilisés par 2-, OH-, -, qui possèdent des orbitales p entièrement remplies, et pas d’orbitales d d’un niveau voisin; exemples: K3[Fe4] avec Fe(+ VI) ou K2[Ni6] avec Ni(+ IV) et K3[Cu6] avec Cu(+ III).RéactivitésLa réactivité des ions complexes est liée à un certain nombre de facteurs évoqués précédemment. Les valeurs des énergies de stabilisation due au champ des ligands (L.F.S.E.), déterminées d’après l’occupation des différentes orbitales d, semblent jouer un rôle important; les complexes octaédriques à bas spin (d6) du Co(III) qui présentent une forte L.F.S.E. sont inertes, comme d’ailleurs ceux du Cr(III). Cependant, il ne faut pas relier réactivité et stabilité ionique; l’ion Co(NH3)63+ est inerte, et il persiste plusieurs heures en solution acide bien qu’il soit thermodynamiquement peu stable puisque l’on a lg K 年 25 pour la réaction [Co(NH3)6)3+ + 6H3+[(Co(H2O)6]3+ + 6NH4+.5. Mécanismes de réactionsDu point de vue cinétique, les complexes inertes présentent des réactions suffisamment lentes pour être étudiées par les techniques conventionnelles. Par contre, pour les complexes labiles (temps de demi-réaction de 10-8 à une seconde), seul le développement de techniques spéciales, telles que les méthodes de compétition, de relaxation ou de perturbation spectroscopiques (R.M.N., R.P.E.) et électrochimiques, en a permis l’étude. Malgré tout l’intérêt présenté par ces techniques, nous nous limiterons aux principaux résultats concernant les mécanismes réactionnels de deux types de réactions importantes des complexes: substitutions et oxydo-réductions. Ces indications sont nécessairement qualitatives et sont simplement des exemples de la façon dont les chimistes tentent d’interpréter les données expérimentales.SubstitutionsCes réactions sont des substitutions nucléophiles (SN) que l’étude cinétique permet de classer en deux groupes:1. Les réactions S1 du premier ordre à mécanisme dissociatif: les études des réactions de substitutions dans les complexes octaédriques d’un ion comme Co3+, par exemple, ont montré que l’étape importante (c’est-à-dire celle qui détermine la vitesse de la réaction) est celle qui implique la rupture de la liaison entre Co3+ et le groupe sortant. Le groupe entrant n’est pas impliqué dans cette étape initiale. La seconde étape, très rapide, est l’introduction du substituant. Exemple:
– classe b . Cations à configuration électronique externe à dix-huit électrons: Cu+, Ag+, Au+, Zn2+, Cd2+, Hg2+... Les caractéristiques sont opposées à celles des ions de classe a, conduisant à des liaisons essentiellement covalentes, donc à des ordres d’affinité inversés.– classe c . De caractère intermédiaire, cette classe comprend notamment les ions des métaux de transition... Fe2+, Co2+, Ni2+, Cu2+.Ce type de notion a été développé par Pearson (1963) dans la théorie des acides (ions métalliques) durs et mous et des bases (coordinats) dures et molles. C’est la théorie H.S.A.B. (hard and soft acid base theory ). La classification correspondante présente de grandes analogies avec la précédente, la classe a représentant les acides durs et la classe b les acides mous. La règle fondamentale stipule que les acides durs préfèrent les bases dures et que les acides mous préfèrent les bases molles. C’est ainsi que les complexes fluorés de l’aluminium seront stables (F- base dure et Al3+ acide dur) alors que les complexes fluorés du mercure seront peu stables (Hg2+ acide mou).CoordinatsNature des atomes donneursDans la théorie H.S.A.B., les coordinats sont aussi classés en bases dures et molles. Une base dure aura d’une façon générale une faible polarisabilité, une grande électronégativité, une charge négative, une petite dimension et formera des liaisons ioniques. C’est le cas, par exemple, de H2O, OH-, -, P43-, S2-4 ... Les bases molles ont des caractéristiques inverses: R2S, RSH, RS-, I-, SC-, (S23)2-...Effets chélate et macrocycliqueLa stabilité des complexes est fortement accrue par chélation et par effet macrocyclique (cf. COMPLEXES - Chimie).Dimension et nombre de cyclesLes cycles pentagonaux les plus stables sont ceux qui ne renferment pas de double liaison (éthylènediamine...); les cycles hexagonaux les plus stables sont ceux qui contiennent une ou deux doubles liaisons ( 廓-dicétones, esters 廓-cétoniques).Plus le nombre de cycles formés est élevé, plus la stabilité est importante; la stabilité croît lorsque l’on passe de NH3 (0 cycle) à l’éthylènediamine (1 cycle), à la diéthylènetriamine (2 cycles), puis à la triéthylènetétramine (3 cycles).Propriétés acido-basiques du coordinatLes ions hydrogène et l’ion métallique sont des acides de Lewis, donc des accepteurs d’électrons. Il y a donc une similitude entre la complexation de l’ion métallique par un coordinat et la neutralisation d’une base par les ions hydrogène. En outre, la plupart des coordinats sont des bases conjuguées d’acides, et on peut penser que, d’une façon générale, plus le pK de la base est élevé, plus la stabilité du complexe métallique est grande. Cette corrélation est souvent vérifiée, mais là aussi il y a de nombreuses exceptions.Influence de substituant(s) sur le coordinatCes substitutions peuvent, d’une part, modifier les propriétés acido-basiques du coordinat, donc la stabilité des complexes, et, d’autre part, induire des effets stériques qui peuvent être importants et largement compenser les variations de stabilité dues aux variations de basicité.RésonancesLes résonances augmentent toujours la stabilité des complexes; pour qu’elles aient lieu, il faut que ceux-ci aient une structure plane ou s’écartant peu de la planéité; l’augmentation du domaine offert aux électrons délocalisés accroît encore la stabilité; c’est le cas des porphyrines ou, mieux encore, des phtalocyanines.Stabilisation de degrés d’oxydation inhabituelsLa coordination permet de stabiliser certains degrés d’oxydation inhabituels.Les degrés d’oxydation les plus petits, éventuellement même négatifs, sont stabilisés par coordination avec CO, C-, RCN, PCl3, la 2-2 -bispyridine; exemples: Na2[Fe(CO)4], qui contient Fe(face=F0019 漣 II), ou K4[Ni(CN)4] et Ni(CO)4, qui contiennent Ni(0). On attribue cette stabilisation à la formation d’orbitales moléculaires du type 神 entre le métal et son coordinat.Les degrés d’oxydation élevés sont stabilisés par 2-, OH-, -, qui possèdent des orbitales p entièrement remplies, et pas d’orbitales d d’un niveau voisin; exemples: K3[Fe4] avec Fe(+ VI) ou K2[Ni6] avec Ni(+ IV) et K3[Cu6] avec Cu(+ III).RéactivitésLa réactivité des ions complexes est liée à un certain nombre de facteurs évoqués précédemment. Les valeurs des énergies de stabilisation due au champ des ligands (L.F.S.E.), déterminées d’après l’occupation des différentes orbitales d, semblent jouer un rôle important; les complexes octaédriques à bas spin (d6) du Co(III) qui présentent une forte L.F.S.E. sont inertes, comme d’ailleurs ceux du Cr(III). Cependant, il ne faut pas relier réactivité et stabilité ionique; l’ion Co(NH3)63+ est inerte, et il persiste plusieurs heures en solution acide bien qu’il soit thermodynamiquement peu stable puisque l’on a lg K 年 25 pour la réaction [Co(NH3)6)3+ + 6H3+[(Co(H2O)6]3+ + 6NH4+.5. Mécanismes de réactionsDu point de vue cinétique, les complexes inertes présentent des réactions suffisamment lentes pour être étudiées par les techniques conventionnelles. Par contre, pour les complexes labiles (temps de demi-réaction de 10-8 à une seconde), seul le développement de techniques spéciales, telles que les méthodes de compétition, de relaxation ou de perturbation spectroscopiques (R.M.N., R.P.E.) et électrochimiques, en a permis l’étude. Malgré tout l’intérêt présenté par ces techniques, nous nous limiterons aux principaux résultats concernant les mécanismes réactionnels de deux types de réactions importantes des complexes: substitutions et oxydo-réductions. Ces indications sont nécessairement qualitatives et sont simplement des exemples de la façon dont les chimistes tentent d’interpréter les données expérimentales.SubstitutionsCes réactions sont des substitutions nucléophiles (SN) que l’étude cinétique permet de classer en deux groupes:1. Les réactions S1 du premier ordre à mécanisme dissociatif: les études des réactions de substitutions dans les complexes octaédriques d’un ion comme Co3+, par exemple, ont montré que l’étape importante (c’est-à-dire celle qui détermine la vitesse de la réaction) est celle qui implique la rupture de la liaison entre Co3+ et le groupe sortant. Le groupe entrant n’est pas impliqué dans cette étape initiale. La seconde étape, très rapide, est l’introduction du substituant. Exemple: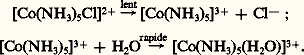 Une caractéristique d’un tel processus est l’influence nulle du groupe entrant sur la vitesse de la réaction. Celle-ci ne dépend que de la concentration du complexe du métal (réaction du premier ordre).2. Les réactions S2 du deuxième ordre (ou bimoléculaires) à mécanisme associatif.Dans les complexes carrés plans, il est possible d’envisager un mécanisme comprenant d’abord le rattachement du groupe entrant avec formation d’un état de transition pentacoordiné. Exemple:
Une caractéristique d’un tel processus est l’influence nulle du groupe entrant sur la vitesse de la réaction. Celle-ci ne dépend que de la concentration du complexe du métal (réaction du premier ordre).2. Les réactions S2 du deuxième ordre (ou bimoléculaires) à mécanisme associatif.Dans les complexes carrés plans, il est possible d’envisager un mécanisme comprenant d’abord le rattachement du groupe entrant avec formation d’un état de transition pentacoordiné. Exemple: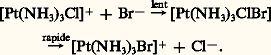 Ce mécanisme associatif (car l’étape déterminant la vitesse de substitution implique l’association du complexe et du groupe entrant) est confirmé par le fait que les vitesses de substitutions des complexes carrés plans dépendent fortement de la nature du groupe entrant (réaction du deuxième ordre).Un processus analogue pour les complexes octaédriques implique la formation transitoire (plus difficile pour des raisons stériques) d’un complexe heptacoordiné.Dans le cas des complexes octaédriques, pour une réaction S2, si l’attaque se fait en position cis par rapport au coordinat qui est éliminé, il n’y a pas transposition; mais cette dernière se produit si l’attaque se fait en position trans :
Ce mécanisme associatif (car l’étape déterminant la vitesse de substitution implique l’association du complexe et du groupe entrant) est confirmé par le fait que les vitesses de substitutions des complexes carrés plans dépendent fortement de la nature du groupe entrant (réaction du deuxième ordre).Un processus analogue pour les complexes octaédriques implique la formation transitoire (plus difficile pour des raisons stériques) d’un complexe heptacoordiné.Dans le cas des complexes octaédriques, pour une réaction S2, si l’attaque se fait en position cis par rapport au coordinat qui est éliminé, il n’y a pas transposition; mais cette dernière se produit si l’attaque se fait en position trans :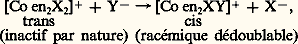 en désignant une molécule d’éthylènediamine: NH2 漣CH2 漣CH2 漣NH2.Réactions d’oxydo-réductionsIl existe deux types de transfert d’électrons. Le transfert électronique d’un complexe à un autre fait intervenir la formation transitoire d’un complexe activé ponté mis en évidence à l’aide d’isotopes radioactifs par Taube et ses collaborateurs. [Co(NH3)536Cl]2+ par exemple est dissous dans une solution contenant Cr2+ et des ions Cl- non radioactifs; après réduction, on obtient [Cr(H2O)536Cl]2+ qui ne contient que du chlore radioactif. Le complexe activé qui permet le transfert d’électrons doit donc comporter un atome de chlore formant un pont permettant le transfert Cr2+ + Co3+Cr3+ + Cr2+. Dans un tel cas, non seulement des électrons, mais aussi des atomes ou des groupes d’atomes sont transférés. C’est le transfert atomique ou mécanisme à sphère interne.Dans d’autres réactions rapides, on envisage un transfert direct entre les deux complexes. Il y a transfert d’électrons dans un complexe issu d’une collision mais sans substitution. Exemple:
en désignant une molécule d’éthylènediamine: NH2 漣CH2 漣CH2 漣NH2.Réactions d’oxydo-réductionsIl existe deux types de transfert d’électrons. Le transfert électronique d’un complexe à un autre fait intervenir la formation transitoire d’un complexe activé ponté mis en évidence à l’aide d’isotopes radioactifs par Taube et ses collaborateurs. [Co(NH3)536Cl]2+ par exemple est dissous dans une solution contenant Cr2+ et des ions Cl- non radioactifs; après réduction, on obtient [Cr(H2O)536Cl]2+ qui ne contient que du chlore radioactif. Le complexe activé qui permet le transfert d’électrons doit donc comporter un atome de chlore formant un pont permettant le transfert Cr2+ + Co3+Cr3+ + Cr2+. Dans un tel cas, non seulement des électrons, mais aussi des atomes ou des groupes d’atomes sont transférés. C’est le transfert atomique ou mécanisme à sphère interne.Dans d’autres réactions rapides, on envisage un transfert direct entre les deux complexes. Il y a transfert d’électrons dans un complexe issu d’une collision mais sans substitution. Exemple: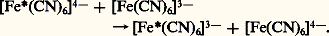 Il a simplement été montré des variations des longueurs de liaisons Fe 漣C lors du transfert électronique. C’est un mécanisme à sphère externe.
Il a simplement été montré des variations des longueurs de liaisons Fe 漣C lors du transfert électronique. C’est un mécanisme à sphère externe.
Encyclopédie Universelle. 2012.